
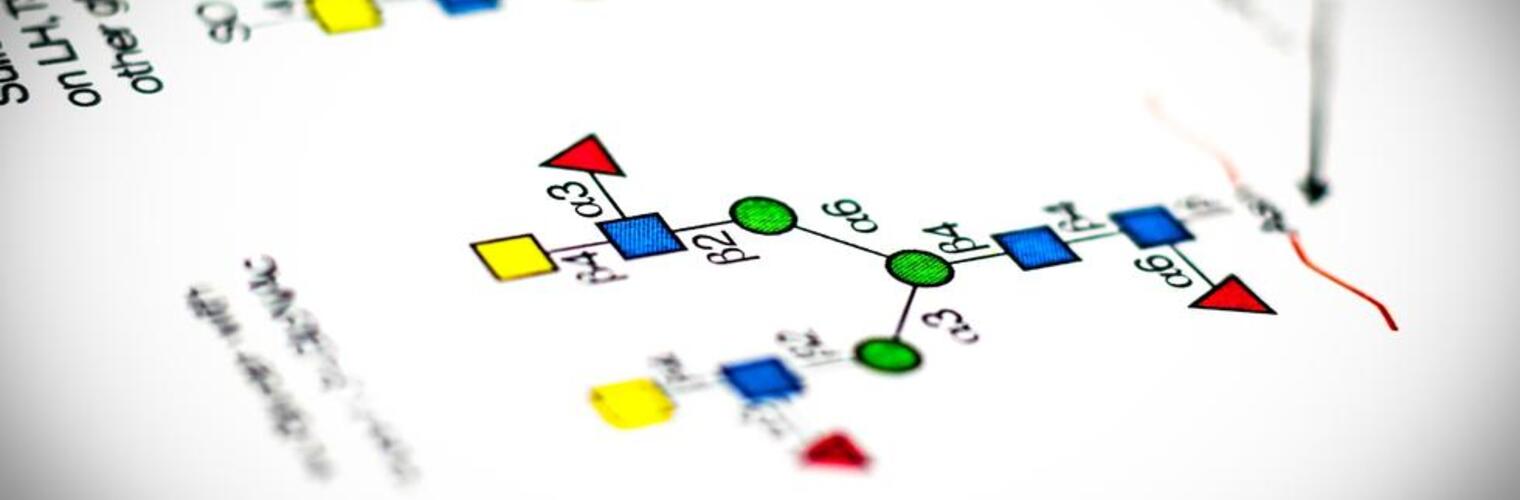


Welcome to the National Center for Functional Glycomics (NCFG) at Beth Israel Deaconess Medical Center, Department of Surgery, Harvard Medical School in Boston, an R24 National and Regional Resource Center, funded by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH). Analogous to Genomics and Proteomics, Glycomics focuses on defining the structures and functions of complex carbohydrates, as found in glycoproteins, glycolipids, and glycosaminoglycans. Complex carbohydrates are important in many physiological processes and alterations in glycosylation are associated with vast numbers of diseases and disorders. The specific focus of the NCFG is technology dissemination and service in the glycosciences with an emphasis on exploring the molecular mechanisms of glycan recognition by proteins important in human biology and disease. We aim to provide assistance and answers to glycoscience questions and problems, so please reach out to us to see how we can help in your research endeavors.
We offer multiple types of glycan and glycopeptide microarrays to the community, much below commercial costs, as a full service- we will analyze your samples and provide you with the data and interpretation. You do not need to be a member of any specific group or organization. Please contact us or make a request for services and we can direct you to the next step to analyze your sample of interest for glycan binding interactions!
The NCFG is affiliated with the CFG, and is funded by the National Institute of General Medical Sciences of the NIH. The Center Director is Richard D. Cummings, Ph.D.
Stats
| Microarrays | Datasets | Protocols |
| 12 | >550 | 18 |